 |
| Hitos en el estudio del ADN antiguo. Luca Ermini et al (2014). |
Hace años se popularizó la llamada Eva mitocondrial (hipótesis Arca de Noé), o ancestro común más reciente para el ADN-mt. Sin embargo, los humanos actuales no provenimos de una sola mujer: Los modelos teóricos y computacionales muestran que número de individuos capaces de reproducirse fue siempre de al menos 4.000. Estos estudios cuentan con un problema importante: la calibración del reloj molecular, del número de sustituciones (mutaciones) por lugar del genoma (sitio) y unidad de tiempo.
La paleogenética, en definitiva, ha aportado un grado muy elevado de certeza al conocimiento de:
La paleogenética, en definitiva, ha aportado un grado muy elevado de certeza al conocimiento de:
- La estructura formada por las diferentes poblaciones humanas en un momento determinado, incluyendo su tamaño.
- La fecha de la separación entre poblaciones (pero DeSalle, 2016).
- La movilidad de estas poblaciones y los eventos de hibridación.
- La adscripción de un fósil a una determinada población, tras su análisis genético.
 |
| Eventos de hibridación humana. |
Carles Lalueza-Fox (2013) repasa algunos problemas de interpretación:
- Los problemas asociados con los relojes moleculares.
- La hipótesis de reloj molecular se basa en la regularidad del proceso de mutación en regiones genéticas neutrales a lo largo de tiempo, implicando así la posibilidad de utilizarlo como un estimador de tiempo para la evolución molecular. Hay, sin embargo, algunos problemas con la exactitud de un reloj molecular, de forma que en cada nueva toma de muestras a menudo se produce un recalibrado.
- La diversidad genética actual (ya sea una población de estudio o de una especie) no está caracterizada en su totalidad. Una inclusión adicional en la muestra, puede atrasar las fechas considerablemente.
- La tasa de mutación es una estimación, con pruebas contradictorias.
- Hay que disponer de fechas exactas, pero el registro fósil no es muy preciso.
- Hay que trabajar con regiones genómicas selectivamente neutras, pero la existencia de elementos reguladores ubicuos y barridos selectivos no detectados hace que su derterminación no sea obvia.
- La diferencia entre la divergencia de secuencias genéticas y la divergencia de especies.
- La teoría de la coalescencia nos permite ir hacia atrás en el tiempo a partir de la variación genética existente hasta encontrar antepasados comunes, lo que proporciona inferencias sobre la demografía de la población y la divergencia genética.
- Los tiempos de coalescencia siempre son anteriores al de divergencia de las especies, debido a que en el momento de la separación existía una cierta variación genética. Estos tiempos serán más cercanos en la medida en que la población ancestral haya sido más pequeña y con ello más próxima genéticamente.
- Las limitaciones de los marcadores uniparentales (ADN-mt y ADN-Y)
- Los marcadores uniparentales han fracasado en la detección de los procesos evolutivos reales: Cuando el tamaño de la población permanece constante durante largo tiempo, el ADN uniparental tiende a aglutinarse en algún momento, de forma que los eventos genéticos anteriores son indetectables.
- Frente al dogma de herencia exclusivamente por vía materna del ADN-mt Luo et al (2018) han informado de múltiples casos de herencia biparental. Este descubrimiento cuestiona seriamente todas las hipótesis sustentadas en la secuenciación de ADN-mt antiguo.
- La forma en que la expresión de un genoma configura el fenotipo (incluyendo la morfología y la cognición).
- Las publicaciones de los genomas humano y del chimpancé no cumplieron las expectativas para la comprensión de la base genética de las diferencias morfológicas (y cognitivas) que existen entre estas dos especies. El problema reside en las dificultades en la comprensión de la función del gen y también en la complejidad del genoma que funciona por encima del nivel del ADN. Existen muchos elementos reguladores que interactúan con redes de genes. Genomas similares o incluso idénticos podrían producir diferentes fenotipos como resultado de diferencias en la regulación de la transcripción de genes.
- Las restricciones físicas y químicas pueden provocar que sólo pueda surgir en el tiempo un determinado conjunto de rasgos adaptativos y que rasgos similares aparezcan de forma independiente en diferentes linajes de homínidos. Diferentes bagajes genéticos pueden producir el mismo fenotipo.
 |
| Individuos con ADN nuclear secuenciado y datación de hace 40 ka o anterior. En azul neandertales, rojo denisovanos y azul HAM. Los asteriscos indican la obtención de genomas en alta cobertura. La interrogación, que se desconoce el sexo. Eel punto azul de Oase 1 indica un ancestro neandertal reciente. Denisova 3, un ancestro neandertal más lejano. Denisova 11 un padre denisovano y madre neandertal. |
ADN antiguos relevantes
- Sima de los Huesos. Meyer et al, 2013.
- Genoma mitocondrial casi completo del Fémur XIII, datado en unos 400 ka.
- Sima de los Huesos. Meyer et al, 2016.
- ADN nuclear a partir del fémur XIII, el incisivo AT-5482, el fragmento de fémur AT-5431, el molar AT-5444 y la escápula AT-6672. ADN-mt de AT-5431.
- Scladina (Bélgica). Peyrégne et al, 2019.
- ADNmt de un maxilar de hembra neandertal (Scladina I-4a) datado por isótopos de uranio y torio en hace 127 ka.
- Más relacionado con Vindija que con Altai.
- Hohlenstein-Stadel (HST; Alemania), Posth et al, 2017. Peyrégne et al, 2019.
- ADNmt de un fémur de macho neandertal hallado en 1937. La datación es incierta. Los métodos genéticos proporcionaron una fecha de hace 124 ka.
- Más relacionado con Vindija que con Altai. HST y Scladina podrían ser miembros de una población neandertal ancestral que dio origen a todos los neandertales secuenciados hasta la fecha, con la excepción del neandertal de Altai. Esta población neandertal ancestral se estableció en el oeste hace unos 120 ka, y los descendientes posteriores puedieron haber migrado al este reemplazando al menos parcialmente a la población oriental de los neandertales representada por el neandertal de Altai.
- Stajnia, Polonia. Andrea Picin et al, 2020.
- Genoma mitocondrial neandertal a partir del molar S5000 hallado en 2007, asociado a teconología micoquiense. Más estrechamente relacionado con Mezmaiskaya-1.
- Chagyrskaya, en los montes Altái. Mafessoni et al, 2020.
- Genoma de alta calidad de una mujer neandertal de hace 80-60 ka, perteneciente a un grupo más relacionado con la población de Vindija que con la de Denisova y más pequeño que cualquiera de éstos.
- Altai, Prüfer et al, 2014.
- Genoma nuclear de alta calidad de una mujer neandertal procedente de la cueva Denisova (Altai), obtenido a partir Denisova 5, un hueso del dedo de un pie datado en 50 ka. Esta cueva es la misma de la que proceden los restos conocidos de “denisovanos”. Además, el estudio incluyó un ADN neandertal a baja cobertura procedente de la Cueva Mezmaiskaya (Cáucaso).
- Denisova, Meyer et al, 2012.
- Genoma nuclear de alta calidad de Denisova 3, un fragmento distal de falange de un infantil hallado en capas de 48-30 ka. En 2010, se había secuenciado el ADN-mt y el ADN nuclear a baja cobertura. También se han obtenido los genomas de Deniosova 2 (Slon et al, 2017), Denisova 4 (Sawyer et al, 2015; Viviane Slon et al, 2015), Denisova 5, Denisova 8 (Sawyer et al, 2015) y Denisova 11, cuyo El ADN-mt corresponde a un neandertal (Brown et al, 2016), pero el ADN nuclear ha revelado que el padre del individuo era denisovano (Slon et al, 2018).
- Ust-Ishim, Fu et al, 2014.
- Genoma de alta calidad procedente de un fémur de varón humano moderno de hace 47,48-42,56 ka, hallado en 2008 en un banco del río Irtysh, Ust-Ishim, Siberia. No es ascendiente de ninguna población actual.
- Vindija,
- Green et al, 2010. ADN-mt completo y ADN nuclear combinado de tres ejemplares neandertales (Vi-33.16, Vi-33.25 y Vi-33.26 datados entre hace 44,45-38,31 ka.
- Prüfer et al, 2017. Genoma de alta calidad de una mujer neandertal (Vi-33.19), datada hace 52 ka.
- Briggs et al, 2009.
- ADN-mt neandertal completo a partir de Vindija (Vi-33.25), Feldhofer 1 y 2, Sidrón 1253 y Mezmaiskaya 1.
- Mateja Hajdinjak et al, 2018
- ADN-mt y nuclear de cinco neandertales que vivieron entre hace 47-39 ka: Les Cottés Z4-1514, Goyet Q56-1, Vindija 87 (hembras), Spy 94a y Mezmaiskaya 2 (machos).
- Vindija 87 resultó pertenecer al mismo individuo que Vindija 33.19.
- La homogeneidad genética de estos ejemplares sugiere un remplazo de la población europea desde refugios en el oeste de Europa o desde Asia.
- Gibraltar. Bokelmann et al, 2019.
- ADN de dos cráneos neandertales parciales: Gibraltar 1, hallado en 1848 en Forbes' Quarry, y Gibraltar 2, hallado en 1926 en Devil's Tower. ADNmt de Gibraltar1. Este individuo resultó ser una hembra mientras que Gibraltar 2 era varón.
- Gibraltar 1 está más relacionado con los neandertales anteriores de Europa y Asia Occidental (Scladina, Hohlenstein-Stadel y Mezmaiskaya) que con los más recientes de España (El Sidrón).
- Oase. Fu et al, 2015.
- ADN-mt y ADN nuclear de Oase 1, un humano moderno datado en hace 41,64-37,58 ka cal.
- No es ascendiente de ninguna población actual. Tiene un ascendiente neandertal, seis generaciones atrás.
- Tianyuan. Fu et al, 2012.
- ADNmt y algunas regiones del ADN nuclear de un ejemplar de humano moderno datado en hace 41,144-39,512 ka.
- Emparentado con los asiáticos y algunos americanos actuales y con Goyet.
- Kostenki. Seguin Orlando, 2014.
- ADN-mt y ADN nuclear de K 14, un esqueleto de humano moderno datado directamente en hace ca. 38,68-36,26 ka cal.
- Emparentado con los europeos actuales.
- Goyet. Fu et al, 2016.
- ADN-mt, ADN nuclear y ADN-Y de Q-116-1, un húmero izquierdo de humano moderno datado en 35,16-34,431 ka.
- Emparentado con los europeos actuales y con Tianyuan.
- Sunghir. Sikora et al, 2017.
- Genoma completo de SI, SII, SIII y SIV, datados en 35,283-29,476 ka.
- Emparentado con los europeos actuales, Kostenki y Vestonice.
- Vestonice. Fu et al, 2016. Mittnik et al, 2016.
- ADN-mt, ADN nuclear y ADN-Y de DV-16, un varón datado en 30,71-29,31 ka.
- Emparentado con los europeos actuales.
- Ostuni. Fu et al, 2016.
- ADN-mt y ADN nuclear de Ostuni 1, una hembra datada en 30,71-29,31 ka.
- Emparentado con los europeos actuales.
- Relativamente próximo a Vestonice.
- Malt'a. Raghavem et al, 2014.
- ADN-mt y ADN nuclear de un individuo juvenil datado en hace 24,52-24,09 ka.
- Ancestro de los actuales europeos y asiáticos.
- Relativamente próximo a los nativos americanos.
- El Mirón. Fu et al, 2016.
- ADN-mt y ADN nuclear de un dedo del pie de una hembra datada en 18,83-18,61 ka.
- Emparentado con los europeos actuales y con Goyet.
- Afontova Gora. Fu et al, 2016.
- ADN-mt y ADN nuclear de una hembra joven datada en 16,93-16,49 ka.
- Relativamente próximo a Malt'a y a los nativos americanos.
- Villabruna. Fu et al, 2016.
- ADN-mt, ADN nuclear y ADN-Y del fémur de un varón datado en 14,18-13,78 ka.
- Emparentado con los europeos actuales. Más cercano a los antiguos pobladores del próximo Este que a los antiguos europeos.
- Bichon. Jones et al, 2015.
- Genoma de un varón datado en 13,77-13,56 ka.
- Emparentado con Villabruna. Conectado con los antiguos pobladores del Este de Asia.
- Satsurbia. Jones et al, 2015.
- Genoma de un varón datado en 13,88-13,13 ka.
Emparentado con los actuales europeos. Ascendencia euroasiática basal. - Anzick. Rasmussen et al, 2014.
- Genoma de un bebé varón (Anzick 1) datado hace ca 12.707-12.556 ka.
- Emparentado con los nativos del centro y sur de América.
- Natufian (Raqefet Cave). Lazaridis et al, 2016.
- ADN-mt y ADN-Y de individuos datados en 11,84-9,76 ka.
- Ascendencia euroasiática basal.
- Upward Sun River. Moreno-Mayar et al (2018)
- Genoma completo de USR1, un bebé de seis semanas, datado hace ca 11,5 ka.
 |
Relaciones entre poblaciones contemporáneas y momento aproximado de divergencia. Las líneas continuas y de puntos indican eventos de mezcla con pruebas más o menos sólidas respectivamente.
ANE: Euroasiáticos del Norte antiguos.
EEF: Agricultores tempranos europeos.
WHG: Cazadores recolectores europeos occidentales.
|
Principales conclusiones de los estudios paleogenéticos
- TMRCA (antigüedad del antecesor común más reciente):
- 1,1 Ma para los haplotipos (cenhaps) de las regiones codificantes del centrómero de los cromosomas 11 y 12; 817 ka para el cromosoma 8 y 700 ka para el cromosoma X (Langley et al, 2019).
- Evidencia la introgresión de una especie arcaica aún desconocida o la supervivencia de un haplotipo antiguo.
- 765-550 ka, del H. sapiens y preneandertales-denisovanos, para el ADNn y ADNmt (Meyer et al, 2016; Fernando L. Mendez et al, 2016; Alan R. Rogers, Ryan J. Bohlender y Chad D. Huff, 2017; Prüfer et al, 2017; Mateja Hajdinjak et al, 2018).
- Esta fecha descarta a ejemplares más jóvenes clasificados en el grado heidelbergensis, como Arago o Petralona, como posibles ancestros comunes a neandertales y HAM, pero no a Homo antecessor.
- 440-390 ka entre neandertales y denisovanos (Meyer et al, 2016; Prüfer et al, 2017; Mateja Hajdinjak et al, 2018). Alan R. Rogers, Ryan J. Bohlender y Chad D. Huff (2017) proporcionan una fecha de hace 744 ka. Rogers et al, 2019, de 731 ka.
- 498-295 ka, entre neandertales y sapiens para el ADN-mt (Fu et al, 2013; Adrien Rieux et al, 2014).
- Según Aida Gómez-Robles (2019), cualquier tiempo de divergencia entre los neandertales y los humanos modernos de menos de 800 ka habría implicado una evolución dental inesperadamente rápida en los primeros neandertales de Sima de los Huesos.
- 370 ka para el ADN-Y de sapiens y neandertales (Petr et al, 2020; Mendez et al, 2016 habían calculado 806-447 ka)
- 363 ka entre denisovanos de Altai y denisovanos que hibridaron con papúes hace 45,7 ka (Jacobs et al, 2019).
- 283 ka entre denisovanos de Altai y denisivonos que hibridaron con paúes hace 29,8 ka (Jacobs et al, 2019).
- 270 ka para el ADNmt conocido de los neandertales (Posth et al, 2017; Peyrégne et al, 2019).
- 260 ka para la divergencia entre cazadores recolectores sudafricanos datados entre hace 2-0,3 ka y otros grupos de sapiens (Schlebusch et al, 2017).
- 254-190 ka para el árbol Y de los humanos actuales (Monika Karmin et al, 2015; Chiara Barbieri et al, 2016; Poznick et al, 2016; Eran Elhaik et al, 2016)
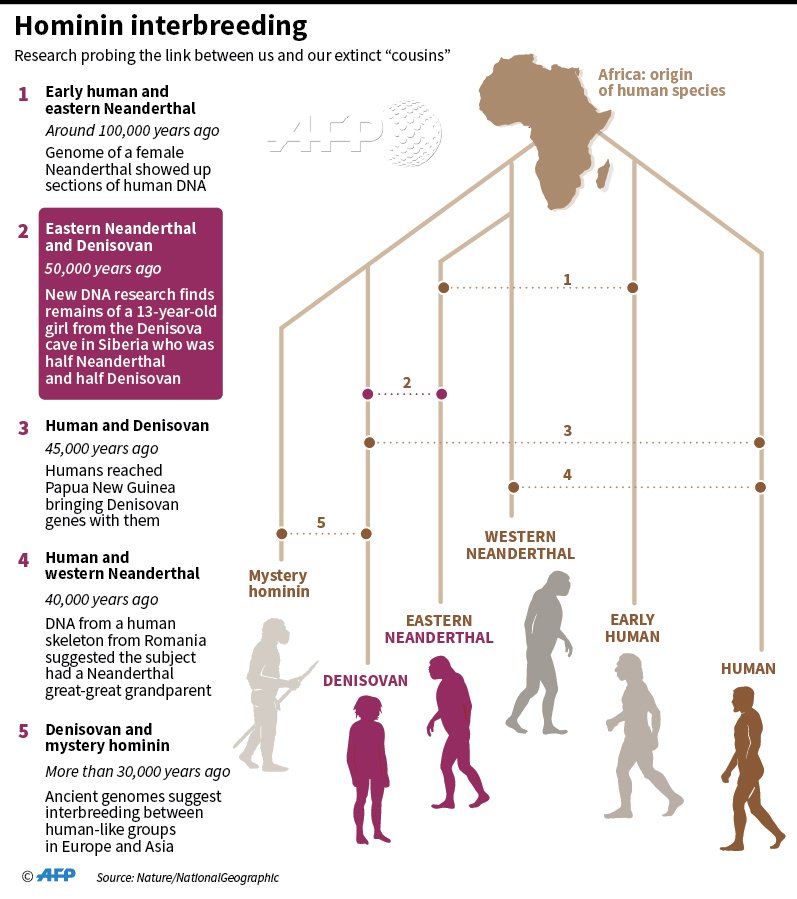
- <200 ka para el árbol del ADNmt de todos los humanos actuales y separación entre los haplogrupos L0 (sur de África) y L1'6 (Centro y Este de África) (Soares et al, 2016; Chan et al, 2019; comentario de José Luis Moreno Garbayo).
- 150-135 para la separación entre los haplogrupos del ADNmt de los sapiens L1 (África Central y Oriental) y L2'6 (África Oriental) (Soares et al, 2016).
- 150-130 ka para la población "Altai Neandertal" y otras poblaciones neandertales. 100-70 ka para la separación entre Vindija Vi-33.19 y otras poblaciones neandertales (Prüfer et al, 2017; Mateja Hajdinjak et al, 2018).
- 100-85 para el surgimiento del haplogrupo L2 de sapiens en África Central/Occidental (Silva et al, 2015).
- 95-62 ka para el ADNmt de los no africanos y los africanos con el haplogrupo L3 (Fu et al, 2013).
- >70 ka para la división de L0 de sapiens en el Sur de África (Rito et al, 2013).
- 72 ka para el ADNmt de los humanos actuales no africanos (Adrien Rieux et al, 2014).
- 40 ka entre europeos actuales y asiáticos actuales (Yang y Fu, 2018).
 |
| Haplogrupos del ADNmt del sapiens. Rito et al, 2019. |
- La población de la Sima de los Huesos (400 ka) estaba estrechamente relacionada por vía materna (ADNmt) con los denisovanos (Meyer et al, 2013). El ADN nuclear, sin embargo, confirmó la pertenencia de esta población al linaje neandertal (Meyer et al, 2016).
- La relación con los denisovanos puede provenir de un mestizaje entre poblaciones antepasadas de ambos o a la existencia de un antepasado común de neandertales, denisovanos y la población de la Sima de los Huesos.
- El ADN-mt de los neandertales, distinto al de los denisovanos y al de los individuos de la Sima de los Huesos, procedió quizá de poblaciones africanas que llegaron a Europa después de hace 430 ka. Estas poblaciones posiblemente trajeron consigo el musteriense (la población de las Sima de los Huesos está asociada al achelense).
- Con fósiles muy escasos y poco informativos, la genómica ha revelado la existencia de un grupo arcaico diferenciado: los denisovanos (Meyer et al, 2012).
- La idea de que todos los humanos modernos comparten un origen africano reciente (en los últimos 250 ka) está apoyada en tres evidencias genéticas:
- La mayoría de los loci genéticos examinados hasta la fecha muestran una mayor diversidad en las poblaciones africanas. Hay una relación lineal entre la distancia genética y la distancia geográfica a África (Ramachandran et al, 2005).
- En los árboles filogenéticos, la separación entre las poblaciones africanas y las no-africanas es la ramificación más antigua.
- La mayoría de los estudios atribuyen fechas recientes para la coalescencia molecular o el momento de separación entre las poblaciones africanas y no africanas.
 |
Principales eventos de hibridación de Homo, indicados por las letras griegas.
X. sapiens africano.
Y. sapiens out of Africa.
N. Neandertal.
D. Denisovano.
S. Población arcaica.
d y xyn ilustran dos diferentes nucleótidos; 0 y 1 representan los alelos derivado y ancestral.
|
- Cuando Green et al, 2010 presentaron el primer borrador del genoma neandertal, encontraron mayor similitud en los genomas de las poblaciones no africanas actuales que en los de las poblaciones africanas. La explicación más parsimoniosa era una hibridación entre neandertales y los ancestros de las poblaciones no africanas actuales. Esta conclusión tentativa ha sido reforzada por numerosos estudios posteriores. Otros estudios han demostrado que los residentes modernos del este de Asia tienen genes neandertales en entre un 12% y un 20% más que los europeos. Villanea y Schraiber (2018) consideran que el escenario más probable es que primero, los neandertales se cruzaron con humanos modernos después de que salieron de África, y luego otra vez, después de que se dividieron en poblaciones europeas y asiáticas orientales. Según los estudios paleogenéticos, entre neandertales, denisovanos, otras poblaciones arcaicas y los primeros humanos modernos se produjeron varios eventos de flujo de genes.
- Entre los ancestros de neandertales y denisovanos y una población arcaica desconocida que se separó del resto hace 2,5-1,7 Ma y tenía un tamaño relativamente grande de 10.000-46.000 individuos (Rogers et al, 2019).
- Entre neandertales y una población africana. Introgresión hace 460-219 ka de una población africana en los neandertales europeos. Sima de los Huesos reveló un ADNmt estrechamente relacionado con los denisovanos (Meyer et al, 2016) que fue reemplazado por un ADNmt más similar al de los sapiens (Posth et al, 2017). Petr et al (2020) sugieren una hibridación hace 300 ka que produjo la sustitución del cromosoma Y de los neandertales, que en los modernos es más próximo al de los sapiens que al de los denisovanos.
- Entre humanos modernos tempranos y neandertales, hace más de 145 ka. Introgresión sapiens en el genoma de Vi-33.19. Prüfer et al, 2017.
- Entre humanos modernos tempranos y neandertales asiáticos, hace ca 100 ka. Introgresión sapiens en el genoma de Denisova 5. Kuhlwilm et al, 2016.
- Los neandertales de las montañas de Altai descendían de una hibridación entre humanos modernos y neandertales hace aproximadamente 100 ka, posiblemente en el Levante Mediterráneo. Esta contribución genética no se detecta ni en los denisovanos ni en los neandertales europeos, lo que sugiere la existencia de al menos dos poblaciones neandertales separadas.
- Estas conclusiones suponen un apoyo sólido a un Out of Africa temprano.
- Entre humanos modernos ancestros de los nativos no africanos actuales y neandertales
europeos, hace ca 55-50 ka, en algún lugar del oeste de Eurasia. Introgresión de los neandertales de Vindija en los sapiens. Green et al, 2010. Las actuales poblaciones humanas no africanas mantienen un 2% de ADN neandertal en su genoma. - La similitud genética de los asiáticos actuales con los neandertales es ligeramente, pero significativamente mayor que la de los europeos. Este patrón es lo contrario de lo que se esperaba por motivos puramente paleoantropológicos, ya que no se han identificado fósiles neandertales en el Este de Asia. Vernot et al (2016) han identificado introgresión neandertal en tres momentos diferentes:
- Un pulso en los no africanos.
- Un segundo pulso en los asiáticos y europeos. Benjamin Vernot y Joshua M. Akey (2014, 2015). Fu, 2015.
- Un tercer pulso en los asiáticos (Jeffrey D. Wall et al, 2013); Benjamin Vernot y Joshua M. Akey (2014, 2015).
- Entre humanos modernos ancestros de los asiáticos del este y del sur actuales y una población desconocida, emparentada con denisovanos y neandertales, después de hace 60 ka. Introgresión de una población desconocida en el sapiens. Mondal et al, 2016. Teixeira y Cooper, 2019.
- Teixeira y Cooper, 2019 sugieren que este evento de hibridación se produjo en la India.
- Entre humanos modernos ancestros de los actuales habitantes de la isla de Flores y una población desconocida. Introgresión de una población desconocida en el sapiens. Teixeira y Cooper, 2019.
- Entre humanos modernos ancestros de la población de Ust-Ishim y neandertales hace 60-50 ka. No hay descendientes actuales de la población de Ust-Ishim (Fu, 2014).
- Entre humanos modernos ancestros de los nativos melanesios y asiáticos del este y los denisovanos hace 54-30 ka. Introgresión de denisovanos cercanamente emparentados con los denisovanos de Altai en los sapiens. Meyer et al, 2012. Vernot et al, 2016. Sankararaman et al, 2016. Browning et al, 2018. Jacobs et al, 2019. Teixeira y Cooper, 2019.
- Sankararaman et al (2016) han localizado introgresión denisovana en menor proporción en los nativos del Himalaya y en el sur y centro de la India. Esta diferencia podría explicarse mediante un único evento de introgresión, seguido de dilución en diferentes grados en las distintas poblaciones o por un mínimo de tres eventos de introgresión.
- Browning et al, 2018, detectaron una segunda oleada de introgresión entre humanos modernos ancestros de los nativos asiáticos del este y los denisovanos. Introgresión de denisovanos lejanamente emparentados con los denisovanos de Altai en los sapiens.
- Jacobs et al, 2019 han identificado tres eventos de introgresión diferentes:
- En papúes, un linaje denisovano hace 45,7 ka.
- En papúes, otro linaje denisovano diferente hace 29,8 ka.
- En asiáticos orientales, un tercer linaje, en fechas más recientes.
- Para Jinam et al (2017); Teixeira y Cooper, 2019, se produjo un evento de hibridación adicional entre los ascendientes de los actuales nativos filipinos y los denisovanos.
- En el ADN de las actuales poblaciones europeas, se detecta un muy pequeño porcentaje de ascendencia denisovana (0,1%; Skov et al, 2020; Bergström et al, 2020). La introgresión denisovana (y neandertal) ha dejado pequeños rastros también en poblaciones subsaharianas (Lu Chen et al, 2020).
- Con independencia de si la ascendencia denisovana procede de una hibridación sapiens denisovana o se introdujo a través de una hibridación con neandertales que se habían mezclado anteriormente con denisovanos, los haplotipos denisovanos presentes en los europeos no proceden de la población de Altai, sino de otra que se separó de la de Altai hace 400-270 ka (Skov et al, 2020), lo cual es consistente con los hallazgos de Jacobs et al, 2019.
- Entre neandertales asiáticos y denisovanos, hace más de 50 ka. Introgresión neandertal en el genoma denisovano. Meyer et al, 2012.
- Denisova 11 es una hembra con padre denisovano, con algún ancestro neandertal, y madre neandertal, de un población diferente al ancestro neandertal del padre, más cercana a neandertales europeos posteriores (Vindija 33.19) que a los neandertales más antiguos que habitaron la misma cueva (Slon et al, 2018).
- Entre humanos modernos ancestros de los africanos actuales y una población desconocida. Introgresión de una población desconocida en el sapiens. Xu et al, 2017; Durvasula y Sankararaman (2018) para los yoruba.
- Entre denisovanos y una población humana que abandonó África hace ca 1 Ma. Introgresión de una población antigua indeterminada en el genoma denisovano. Meyer et al, 2012. Rogers et al, 2019.
- Lu Chen et al, 2020 han detectado haplotipos neandertales en poblaciones del norte del África subsahariana. Este ADN neandertal procede de dos fuentes:
- Una migración de humanos modernos eurasiáticos a África, en los últimos 50 ka.
- Haplotipos procedentes de poblaciones africanas arcaicas que pasaron a neandertales antes de hace 100 ka y que llegaron a los subsaharianos quizá como resultado de múltiples dispersiones y mezclas entre HAM y neandertales.
- Se han identificado algunas regiones del ADN y el ARN con selección positiva:
- Es notable la incidencia en el sistema inmunitario (p. ej. Gittelman et al, 2016; Sams et al, 2016; Racimo et al, 2016; Dannemann, Andrés y Kelso, 2017; Taskent et al, 2017; Browning et al, 2018; Enard y Petrov, 2018; Jacobs et al, 2019; Greenbaum et al, 2019).
- La adaptación de los tibetanos a condiciones hipóxicas proviene posiblemente de grupos denisovanos (Jeong et al, 2014; Huerta-Sánchez et al, 2014).
- Los bloques genómicos de ascendencia neandertal están enriquecidos en los genes que afectan a los filamentos de queratina (Fu et al, 2014; Vernot y Akey, 2015).
- Los alelos neandertales en los cromosomas 1 y 18 están asociados con una globularidad endocraneal reducida. Estos alelos influyen en la expresión de UBR4 y PHLPP1, dos genes que están implicados en la neurogénesis y la mielinización, respectivamente (Gunz et al, 2018).
- Centrómero del cromosoma 11, relacionado con los receptores del olor (Langley et al, 2019).
- Una de cada tres hembras europeas heredaron el receptor de progesterona neandertal, relacionado con la fertilidad (Zeberg, Kelso y Pääbo, 2020).
- Los humanos actuales con la proteína del canal de sodio Nav1.7 heredada de los neandertales, que presenta tres sustituciones de aminoácidos (M932L, V991L y D1908G), presentan una mayor sensibilidad al dolor (Zeberg et al, 2020).
- En general, tanto la introgresión neandertal como la denisovana, fueron perjudiciales para el HAM, afectando especialmente a la fertilidad, al cerebro y al cerebelo. Se ha planteado la posibilidad de que sapiens haya desarrollado una respuesta inmune contra el cromosoma Y neandertal, de forma que los embarazos de madre sapiens y padre neandertal terminarían en abortos.
- David Gokhman et al, 2014
- Sriram Sankararaman et al, 2014
- Ekaterina E. Khrameeva et al, 2014
- Ivan Juric, Simon Aeschbacher y Graham Coop, 2015
- Kelley Harris y Rasmus Nielsen, 2015
- Charlotte Jane Houldcroft y Simon Underdown, 2015
- Michael Dannemann (presentación de 2015)
- Matthieu Deschamps et al, 2015
- Qiaomei Fu et al, 2016
- Vernot et al, 2016
- Sankararaman et al, 2016
- Simonti et al, 2016
- Dannemann, Andrés y Kelso, 2016
- Fernando L. Mendez et al, 2016
- Harris y Nielsen, 2016
- Ville N. Pimenoff, Cristina Mendes de Oliveira e Ignacio G. Bravo (2016)
- Nédélec et al, 2016
- Quach et al, 2016
- Nuttle et al, 2016
- Ivan Juric, Simon Aeschbacher y Graham Coop, 2016.
- Rajiv C. McCoy, Jon Wakefield y Joshua M. Akey. 2017.
- Prüfer et al, 2017
- Zeberg y Pääbo, 2020.
- Algunas variantes genéticas en los humanos actuales, heredadas de los neandertales, representan la condición del antepasado común (Capra, annual meeting of The American Society of Human Genetics, 2017).
- Hay evidencia de una reducción en el tamaño de la población que se produjo hace algún tiempo antes de hace 1 Ma.
- Tras la separación entre los linajes neandertal-denisovano y sapiens, el tamaño del primero fue muy pequeño durante los primeros 10 ka. Luego se dividió en dos poblaciones regionales, los neandertales y los Denisovanos. La población neandertal creció mucho y se separó en grupos locales aislados, pero luego redujo su tamaño (Alan R. Rogers, Ryan J. Bohlender y Chad D. Huff, 2017; Prüfer et al, 2017; Rogers et al, 2019).
- La población ancestral de los HAM aumentó de tamaño, mientras que las poblaciones ancestrales de Altai y denisovanos disminuyeron aún más en tamaño (Prüfer et al, 2014). Tras la separación de euroasiáticos y africanos, los primeros experimentaron un cuello de botella y se dividieron en poblaciones regionales. La diáspora eurasiática moderna parece haber sido precedida por otra anterior en más de medio millón de años (Alan R. Rogers, Ryan J. Bohlender y Chad D. Huff, 2017).
- Según Sebastian Lippold et al (2014), las diferencias genéticas entre las poblaciones humanas a escala global son mayores para el NRY que para el ADNmt. La población ancestral fue muy pequeña en tamaño (<100) para el Out of Africa. El ratio entre la población reproductora femenina y la población reproductora masculina (Nf/Nm) ha sido mayor que uno a través de la historia de los humanos modernos y se ha incrementado recientemente debido un crecimiento mayor en Nf.
- Sólo 96 genes son diferentes entre los humanos modernos y los neandertales. Algunas de las diferencias génicas corresponden a respuestas del sistema inmune y al desarrollo de las neuronas en el neocórtex del cerebro humano, incluyendo el desarrollo fetal. El gen RB1CC1, está involucrado en la construcción de los huesos, los cartílagos y músculos del sistema musculoesquelético. Las diferencias en regiones no codificantes, posiblemente reguladoras de genes, son más de 3.000 (Prüfer et al, 2014).
- Gokhman et al (2019) han utilizado los patrones de metilación del ADN para inferir la morfología de los denisovanos.
Recursos web
Mapa interactivo con los estudios sobre el ADN humano antiguo.
Bibliografía:
- Luca Ermini et al. 2015. Major transitions in human evolution revisited: A tributeto ancient DNA.
- Montgomery Slatkin y Fernando Racimo. 2016. Ancient DNA and human history
- Bastien Llamas, Eske Willerslev y Ludovic Orlando. 2016. Human evolution: a tale from ancientgenomes
- Rasmus Nielsen, Joshua M. Akey, Mattias Jakobsson, Jonathan K. Pritchard, Sarah Tishkoff y Eske Willerslev. 2017. Tracing the peopling of the world through genomics.
- Yang y Fu. 2018. Insights into Modern Human Prehistory Using Ancient Genomes.
- María Martinón-Torres. 2019. What We have Learned over the Last Decade.
No hay comentarios:
Publicar un comentario